
Archivo BAM: definición e importancia en bioinformática
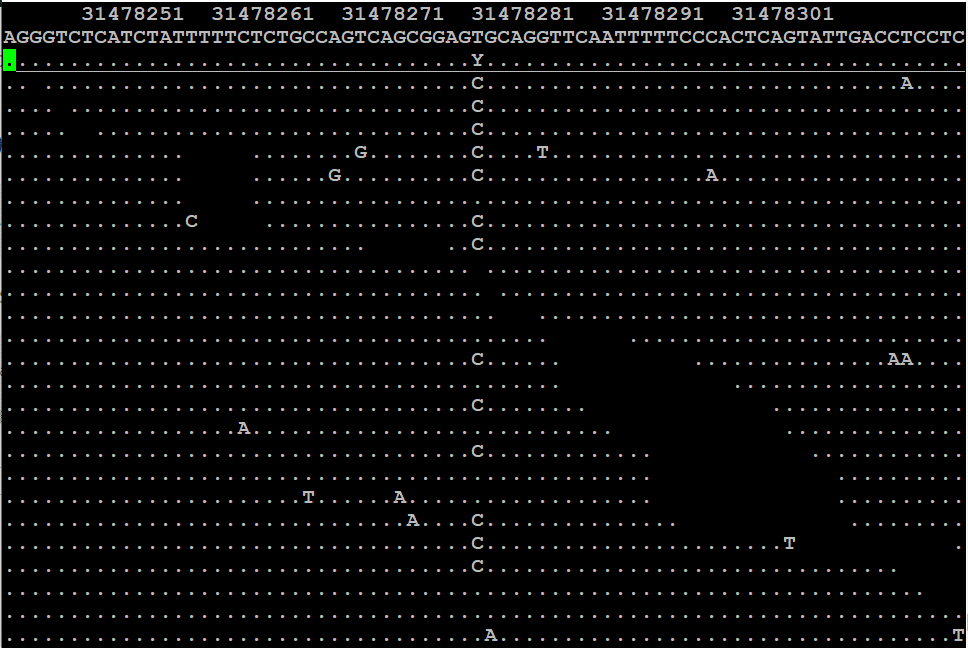
En el mundo de la **bioinformática**, el manejo de datos genómicos se ha convertido en una de las tareas más cruciales para la investigación y el avance en el campo de la biología molecular. Un aspecto fundamental de este manejo de datos es la forma en que organizamos y almacenamos la información genética. Aquí es donde los archivos BAM entran en juego, ofreciendo una forma eficaz de almacenar datos de secuenciación de alto rendimiento. Comprender los **archivos BAM** y su relevancia no solo es vital para investigadores y científicos, sino también para estudiantes y profesionales que se inician en el campo de la bioinformática.
Este artículo está diseñado para proporcionar una visión completa sobre la definición de los archivos BAM, su estructura, funcionamiento y la importancia que tienen en el análisis de datos genómicos. A medida que avanzamos, exploraremos los distintos componentes que conforman estos archivos y cómo son utilizados en diversas aplicaciones de la bioinformática. Al final de este recorrido, no solo estarás familiarizado con los términos técnicos, sino que también comprenderás cómo los archivos BAM impactan la investigación genética y qué avances están llevando a cabo en este ámbito.
Índice
¿Qué es un archivo BAM?
Un archivo BAM (Binary Alignment/Map) es un formato de archivo comprimido que se utiliza principalmente para almacenar datos de alineamiento de secuencias de ADN. Este formato es particularmente útil en el campo de la **secuenciación de nueva generación** (NGS), donde grandes volúmenes de datos son generados. El archivo BAM es la versión binaria de un archivo SAM (Sequence Alignment/Map), lo que significa que contiene la misma información estructural pero de forma más compacta y eficiente.
La **importancia** de los archivos BAM radica en su capacidad para manejar información de alineación de contigs o lecturas de secuenciación frente a un genoma de referencia. Esto permite a los investigadores observar cómo se alinean las secuencias secuenciadas y facilita el análisis posterior, como la detección de variantes genéticas, la identificación de mutaciones y el análisis de expresión génica. Los archivos BAM son comprimidos y, por tanto, ocupan menos espacio, lo que es fundamental cuando se trabaja con datos de secuenciación masiva.
Estructura de un archivo BAM
Los archivos BAM están compuestos por varias secciones clave que contienen importantes datos sobre las secuencias alineadas. Cada interfaz de alineamiento incluye un encabezado y uno o más registros de alineamiento. El encabezado proporciona información general sobre el archivo, como el nombre del genoma de referencia, la longitud de las secuencias y otros parámetros que pueden influir en el análisis.
Por otro lado, los registros de alineamiento contienen información detallada sobre cada una de las lecturas. Cada registro incluye una serie de campos como la identificación de la secuencia, la posición de alineamiento en el genoma de referencia, el indicador de la calidad de la alineación y algunos indicadores sobre las características de las lecturas, como las secuencias de calidad y los **códigos de posicionamiento**. Estos campos permiten a los investigadores evaluar la precisión de la alineación y la calidad de los datos obtenidos.
La diferencia entre archivos BAM y SAM
La principal diferencia entre un archivo BAM y un archivo SAM radica en el formato y la forma en que se almacenan los datos. Mientras que un archivo SAM es un formato de texto plano, lo que significa que puede ser leído y editado fácilmente, un archivo BAM está en formato binario, lo que lo hace más eficiente para el almacenamiento y el procesamiento. Esta **eficiencia** se traduce en tiempos de carga y análisis más rápidos, lo que es esencial cuando se trabaja con los enormes volúmenes de datos generados por la **secuenciación de nueva generación**.
A través de la compresión de datos, los archivos BAM reducen significativamente el espacio de almacenamiento necesario. Este ahorro es crucial en entornos de investigación, donde los datos pueden acumularse rápidamente y la capacidad de almacenamiento se convierte en un problema crítico. Sin embargo, el formato binario también implica que los archivos BAM no son fácilmente legibles por humanos, a diferencia de los archivos SAM que pueden ser inspeccionados y editados manualmente.
Aplicaciones de archivos BAM en bioinformática
Los archivos BAM tienen múltiples aplicaciones en el ámbito de la bioinformática, muchas de las cuales son esenciales para el desarrollo de soluciones innovadoras en la investigación biomédica. Uno de los usos más destacados es en la **detención de variantes genéticas**. Los investigadores utilizan los archivos BAM para comparar secuencias alineadas con un genoma de referencia en búsqueda de **mutaciones, inserciones o deleciones** que pueden estar asociadas con enfermedades genéticas o trastornos.
Además de la detección de variantes, los archivos BAM son utilizados en la cuantificación de la expresión génica. Esto permite a los investigadores entender con mayor precisión cómo las células responden a ciertos tratamientos o condiciones ambientales. Al analizar las lecturas de RNA-Seq almacenadas en archivos BAM, se pueden obtener datos sobre qué genes están siendo expresados y en qué medida, dando información valiosa sobre la biología celular.
Herramientas para trabajar con archivos BAM
Existen diversas herramientas y programas diseñados para trabajar eficazmente con archivos BAM. Entre ellos, **samtools** es una de las más populares y utilizadas en la comunidad de bioinformática. Esta herramienta permite a los usuarios realizar una variedad de operaciones sobre archivos BAM, como visualizar, convertir y manipular los datos de alineación. Gracias a su funcionalidad, samtools es vital para el análisis de datos de secuenciación y se ha convertido en un estándar en el campo.
Otra herramienta destacada es **IGV** (Integrative Genomics Viewer), que permite a los investigadores visualizar de manera gráfica los datos contenida en un archivo BAM. **IGV** proporciona una interfaz fácil de usar que muestra la alineación de lecturas sobre un genoma de referencia, facilitando la interpretación de los resultados y proporcionando una plataforma efectiva para la exploración de datos genómicos.
Desafíos en el manejo de archivos BAM
A pesar de sus múltiples ventajas, trabajar con archivos BAM también presenta ciertos desafíos. Uno de los problemas comunes es tratar con la calidad de los datos obtenidos. Si bien los archivos BAM son útiles para almacenar y analizar datos de alineación, los errores durante la secuenciación pueden llevar a resultados inexactos. Esto requiere que los investigadores implementen técnicas de evaluación de calidad para asegurarse de que los datos utilizados en su análisis sean lo más fiables posible.
Además, la interpretación de los datos derivados de archivos BAM puede ser compleja. Esto implica que los investigadores deben tener una sólida comprensión tanto de la biología como de las herramientas bioinformáticas necesarias para extraer conclusiones válidas de los datos. Por ello, la formación continua y la experiencia práctica son fundamentales para afrontar estos desafíos adecuadamente.
El futuro de los archivos BAM en bioinformática
El futuro de los archivos BAM en bioinformática parece prometedor. A medida que avanzan las tecnologías de secuenciación, también lo hacen las técnicas para el manejo y análisis de los datos. Esto implica que no solo se espera que los archivos BAM evolucionen, sino que también surjan nuevos formatos y herramientas que mejoren la eficiencia y la efectividad del análisis genómico.
Existen investigaciones en curso sobre la **mejora de la compresión de datos** y la eficiencia del almacenamiento, lo que sugiere que los archivos BAM seguirán desempeñando un papel crucial en el análisis de datos de próxima generación. Estas innovaciones son importantes no solo para la investigación genómica, sino también para las aplicaciones clínicas, donde se busca integrar los datos genéticos en la práctica médica cotidiana.
Conclusión
Los archivos BAM son un componente esencial en el campo de la **bioinformática**, fundamental para la organización y análisis de datos secuenciales. A través de su capacidad para almacenar información de alineación de forma eficiente, estos archivos permiten a los investigadores detectar variantes genéticas y analizar la expresión génica. A pesar de los desafíos inherentes al manejo de datos de secuenciación, las herramientas disponibles y la evolución continua de las tecnologías proponen un futuro brillante para el uso de archivos BAM. Comprender la naturaleza y la funcionalidad de los archivos BAM no solo es esencial para aquellos en el ámbito de la bioinformática, sino también crucial para avanzar en la investigación genética y bioasanitaria en general.
Si quieres conocer otros artículos parecidos a Archivo BAM: definición e importancia en bioinformática puedes visitar la categoría Datos.
Guía completa del análisis de metadatos: procesos y técnicas

Cómo se utilizan los datos en la genómica comparativa

Cuáles son los datos de comportamiento de proteínas

Qué son los datos de interacción proteína-proteína

Uso de medicamentos: qué son y su importancia en la salud

Cómo se llevan a cabo análisis de dúplex de datos
Deja una respuesta